�������ڣ�2017��10��19�� �Ķ���2386
ȫ����ҩ�з�������̱��¼�����Щ?���ľʹ˽��м�������

Ŀ ¼
һ����������
1.Naldemedine-����������
2.Abemaciclib-����������
3.��״��������(Shingrix)-����ô���
4.Ustekinumab-FDA��������Ӧ֤��Ⱥ
���������걨���������������ʸ���֤
1.DAS181-��ͻ�����Ʒ���֤
2.Osimertinib-����Ӧ֤��FDAͻ�����Ʒ�����֤
3.Gilteritinib-��FDA��ͻ�����Ʒ���֤
4.Uliprisnilacetate-NDA������
5.Tebipenem pivoxil-��QIDP��֤
6.Pegfilgrastim��������ҩ(Mylan&Biocon)-�յ�FDA��CRL
7.Voretigeneneparvovec-��FDA**ίԱ��һ���Ƽ�
8.���̫������Ƭ(DSUVIA)-�յ�FDA��CRL
9.Apalutamide-�ύNDA
10.Eryaspase-���µݽ�MAA
11.Rucaparib-��FDA�ύsNDA
�����ٴ�����
1.PC-mAb������a���ٴ�����
2.Tinostamustine������/�����ٴ�����
�ġ���ҵ��Ϊ
1.KVD-001-����Ȩת��
2.INB03-����Ȩת��
2017��10��9����10��15��
һ ��������
1.Naldemedine-����������
����Thomson Reuters Integrity���棬���ձ���Ұ����ҩ��ʽ���翪���Ħ�-��Ƭ�������-��Ƭ����˫������Naldemedine 0.2mgƬ(��Ʒ��Symproic)�ڽ������������С���ǰ��Symproic��2017��3�»�FDA���������Ʒǰ�����ʹ������ʹ�ð�Ƭ����ʹҩ������ı��أ���MAA�д���EMA�����Ρ�
��Ұ����ҩ��Symproic��FDA��ʱ�����Ĺ���Ԯ��������ݳƣ������ǰ�Ƭ��ҩ��������ҩ����(���������������Էǰ�����ʹ)�����µ�*������һ�ֲ�����Ӧ���ǰ��Լ��������У���Ƭ��ҩ�����±��صĻ�����Լ��40-50%֮�䡣
Naldemedineҩ����Լױ������Σ��ṹʽ����ͼ��ʾ��

Thomson Reuters Integrity��������������Naldemedine�йص�����ר�����ֱ��ǣ�
������ר����US8536192�����й�ͬ��ר��CN200680027118.7�ѻ���Ȩ������2026��5�µ��ڡ�
����ר����US9315512�����й�ͬ��ר��CN201180064907.9(ԭ��)��CN201510332439.9(�ְ�)���ѻ���Ȩ��ͬʱ����2031��11�µ��ڡ�
����/�����ר����US2015216804����δ�����й���
2.Abemaciclib-����������
����Thomson Reuters Integrity���棬������˾�������걨���ɹ�������Pim-1��ø��CDK6��CDK4���Ƽ�Abemaciclib�ڽ������������С���ǰ��FDA��2017��9��28����Abemaciclib 50mg��100mg��150mg��200mgƬ(��Ʒ����Verzenio)���Ϸ�ά˾Ⱥ����������ǰ�����ڷ����Ʒ��������չ��HR-����/HER2-���Ե����ڻ�ת�������ٰ����ߣ�ͬʱ��������Ӧ֤���е�����ҩ����������ǰ�����ڷ����Ʒ������չ��HR-����/HER-���Ե����ڻ�ת�������ٰ������Լ���ǰ���û��ƺ����չ��HR-����/HER2-���Ե�ת�������ٰ����ߡ�Abemaciclib��FDA��palbociclib(��Ʒ��Ibrance������˾)��ribociclib(��Ʒ����Kisqali��ŵ����˾)֮�����ĵڸ������������ٰ���CDK���Ƽ������е�Ibrance��2015��2�»�FDA������2016���ʵ����21.35��Ԫ��������۶
FDA������Abemaciclibʱ�����Ĺ���Ԯ�����Ұ�֢�о�Ժ�����ݳƣ�2017������������25.271�������ٰ����ߣ�����4.061�������������ٰ�������Լ72%���ٰ���������HR-������HER2-���ԡ�
Abemaciclibҩ��������Σ��ṹʽ����ͼ��ʾ��

�й�ר��CN200980151778.X��/��Ȩ˵�����Ȩ��Ҫ��9�Ի�ѧ�ṹʽ����ʽ������Abemaciclib�����ҩ���Σ�Ȩ��Ҫ��10���÷���������������Ŀ�ҩ������Ϊ�����Ρ�
3.��״��������(Shingrix)-����ô���
������ʷ�˹�˾�ڵ���ʱ��2017��10��13�շ�������ƣ����ô���������������ǰ���˸ù�˾�Ĵ�״��������(Shingrix)����Ԥ�������50����Ⱥ�Ĵ�״�������Ⱦ��Shingrix��һ�ַǻ��ǵ�λ���磬�����һ�ֿ�ԭ�ǵ���E������ϵͳAS01B��������ϵͳּ�ڲ���ǿ���־õ����߷�Ӧ���ɰ����˷��������������������½���Shingrix��������50�꼰������Ⱥ��Ԥ����״����䲢��֢��Ŀǰ����������������ŷ�ˡ����ôĴ����ǡ��ձ��ļ��������ڽ����С���������������罫ͨ��2����ע(2�����߳���0�º�2��)�������߽��֡���ǰ����ʿ����Ԥ�⣬Shingrix���к�������۷�ֵ��ͻ��10����Ԫ��ء�
��������GSK��2016�����FDA�ύ��Shingrix��������Ʒ��������(BLA)����BLA���ύ���ǻ���һ��ȫ���III���ٴ���Ŀ������Ŀ�漰18������(�����ձ�)����37000�������ߣ�������Shingrix����Ч�ԡ���ȫ�Ժ�/������ԭ�ԡ����У�ZOE-50�о�(NCT01165177)��16160��50�꼰��������Ⱥ���п�չ��ZOE-70�о�(NCT01165229)��14800��70����������Ⱥ���п�չ��������ʾ��Shingrix�����ܹ���Ч���ʹ�״����ķ����ʣ������ܹ����ʹ�״���������ʹ(PHN)�����巢���ʣ���������������Ѿ���Ⱦ��״����Ļ���Ҳ����һ������Ч��PHN��һ���ɴ�״���������������ʹ���Ǹ�Ⱦ��״�����ĵ���֢״��2017��9����Ѯ��FDA�����缰�����Ʒ����ίԱ����ȫƱͨ����ͶƱ�������ʾ�Ͽ�Shngrix����Ч���밲ȫ�ԡ�
��״����(shingles)����ˮ��-��״�����(VZV)����ļ��Ը�Ⱦ��Ƥ���������ͱ���ΪƤ����ڲ������������ԣ���Ⱦ��ɳ���DZ���ڼ�������ڵ���Ԫ�ڣ����ֿ������»����ۡ���Ⱦ����ðʱ���������ٴ�������ֳ����������ά����Ƥ����ʹ���ַ�����Ƥ������ǿ�ҵ���֢��Ƥ��һ���е����ԺͰ��ڶηֲ����ص㣬�м����Ե�������ɣ���������ʹ;����������ʹ���ء��ò��÷��ڳ��ˣ�������������**������������������֢������̡���������֢���̷��Ը�Ⱦ������Ժʹ�״���������ʹ(PHN)��
�ݹ��ƣ�������ÿ����100������״��������ò��������ڱ�����ŷ�ޡ���̫�������ơ��������Լ������������½�����Ⱥ����Ⱦ��״������ķ���*�ߡ�����������ҵ����ݱ������г���90%�ij����˰��д�״������գ�50��������Ⱥ��Ⱦ��״��������������ߡ�
4.Ustekinumab-FDA��������Ӧ֤��Ⱥ
ǿ����˾�ڵ���ʱ��2017��10��13�շ�����Ϣ�ƣ�FDA��ǰ���˸ù�˾��Ustekinumab(��Ʒ����Stelara)�������������12�������ù��ƻ�ȫ���Ʒ���������߿�״��м�����ߡ�
Ustekinumab��һ���������-12��-23������*����2009��9�»�FDA���������������18���������ڹ��ƻ�ȫ���Ʒ��ij���߿�״��м�����ߡ�ǿ���Ĺ���ָ����Ustekinumab������������ʼ������ÿ��ֻ���ҩ4�Σ��Ӷ���ΪƤ����ҽ�����仼�ߵ���ѡҩ�
ǿ����˾���乫���л�Ԯ�����ݳƣ���м����һ�����ԡ����������Ե���֢�������ɵ���Ƥ��ϸ���Ĺ�����ֳ��Լ750�������˻�����м��������80~90%Ϊ�߿�״��м�����ߡ����⣬��м�������У�80%��������жȣ���20%�Ļ��߳������ضȵİ߿�״��м����������İ߿�״��м�����ͷƤ���沿���Ӷ������������罻��������Ӱ�졣
Ustekinumab��Thomson Reuters������������14����֮��ص�ר��������*�����US6902734�����й�ͬ��ר��CN01816961.9�����أ��������ķְ�����200810100025.3�ѻ���Ȩ������Ȩ˵�����Ȩ��Ҫ��1�����ˡ�һ�ַ���Ŀ�-IL-12���壬�����SEQIDNO:7��ʾ���������е������ɱ���(VH)��SEQIDNO:8��ʾ���������е������ɱ���(VL)����������Ϊ�˿��塱����Ч�ڽ���2021��8�¡�
�� �����걨���������������ʸ���֤
1.DAS181-��ͻ�����Ʒ���֤
�ܲ���������ʥ���Ǹ��Ansun BioPharma��˾�ڵ�ʱʱ��2017��10��11�շ�������ƣ��ù�˾�������������º������������в�����Ⱦ����ҩDAS181��FDA�϶�Ϊ��ͻ�����Ʒ�����
DAS181�����ô˾����Ʊ��õ��������ںϵ��ף������ðе��Ǵ���������������ڵ���Һ�����壬�ֺ�����������Ⱦ����ʱ����������ϣ���DAS181��ͨ���ѽ���Һ���������Ϻ���������������ĸ�Ⱦ���о�֤ʵ��DAS181��������������Ҫ�ĺ���������-���в���(IFV)�������в���(PIV)��ƫ�β���(MPV)�Լ��˳�������-68(EV-68)���п��������á�DAS181���ص����û���ʹ�ò�����������Һ���������������������ҩ�ԣ����ٴ�ǰ���ٴ��о���Ҳδ�۲쵽��ҩ�Ի���м�������ҩ�ԡ�
Ansun BioPharma��*�������һ��DAS181�����������������סԺ����PIV��Ⱦ��2���ٴ����飬����3���ٴ������Ѵ��ڳﱸ�Ρ����⣬��Ansun BioPharma��FDA�ĺ����£�����130����������˻���ͨ����ͬ����ҩ������������DAS181�����ơ�
�������ר�����ԣ�Thomson ReutersIntegrity����������¼����DAS181��ص�*��ר����US8512710(����Ȩ����Ϊ2002��11��22��)����ר�����й�ͬ��ר��(ԭ��)CN200380107241.6�ѻ���Ȩ����Ч�ڽ���2023��11�£����зְ�����CN200910180019.8����Ϊ���ء�
2.Osimertinib-����Ӧ֤��FDAͻ�����Ʒ�����֤
��˹������˾�ڵ�ʱʱ��2017��10��9�շ�������ƣ��ù�˾���������۵ĵ�����EGFR�Ұ��ἤø���Ƽ�Osimertinib(Tagrisso)����ת����EGFRͻ��-���Է�Сϸ���ΰ�(NSCLC)��һ�����Ƶ���Ӧ֤��FDA��ͻ�����Ʒ���֤����Ϊ�˴�ͻ�����Ʒ���֤֮�����Ģ���FLAURA��������ǰδ���ܹ����Ƶľֲ����ڻ�ת����EGFRͻ������NSCLC����Ϊ���Ƚ���Tagrisso����Ұ��ἤø���Ƽ�(TKI)����Ч���밲ȫ�ԡ�������֣�Tagrisso���TKI(�������������)����λ��չ�����ڷֱ�Ϊ18.9������10.2���¡�����������ת�ƶ����Ȼ��ֵĸ����黼����۲쵽�и��Ƶ������ٴ�������Tagrisso���������ã��䰲ȫ����������ǰ�۲쵽�Ľ��һ�¡�
Ŀǰ�����а���������ŷ�ˡ��ձ����й����ڵ�50������һ������Osimertinibÿ���ҩһ�ε�40mg��80mgƬ����������EGFRT790Mͻ����������NSCLC�������ٴ��������ڿ���Osimertinib���ڸ������ƻ���������������ҩʱ����Ч���밲ȫ�ԡ�
Osimertinibҩ��������Σ��ṹʽ����ͼ��ʾ��
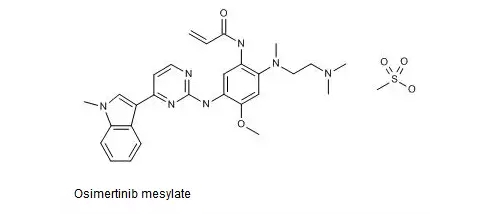
�й�ר��CN201280033773.9��/��Ȩ˵�����Ȩ��Ҫ��1���Ի�ѧ������ʽ������Osimertinib�����ҩ���Σ�Ȩ��Ҫ��2������Osimertinib����Ȩ��Ҫ��3���һ��������Osimertinib�ļ����Ρ�
3.Gilteritinib-��FDA��ͻ�����Ʒ���֤
�ܲ�λ���ձ������İ�˹̩����ҩ�ڵ���ʱ��2017��10��11�շ�������ƣ��ɸù�˾������������Я��FLT3ͻ�����Եķ����Ի������Լ���������Ѫ��(AML)����ҩGilteritinib��FDA�϶�Ϊ��ͻ�����Ʒ������Ӷ�Ϊ������ٴ��������������д�����������������
��˹̩�����乫����Ԯ��������֢Э������ݳƣ�����ÿ��Լ����2.1����AML����������1������Լ1/3��AML���ߴ�������*������FLT3ͻ�䣬��FLT3�ڲ�������������FLT3�Ұ��ἤø�ṹ������Gilteritinib�Դ����ֵ����������������á����⣬Gilteritinib�Ծݱ����������������ܵ�AXLҲ���������á�
Ŀǰ����˹̩����˾���ڽ���4������ٴ����飬����Gilteritinib�ڶ���AML�����е���Ч���밲ȫ�����м��������Է�����/������FLT3+AML����Ϊ�����ADMIRAL���顣
Gilteritinib�Ľṹʽ����ͼ��ʾ��

�й�ר��CN201080020181.4����Ȩ˵�����Ȩ��Ҫ��14�Ի�ѧ������ʽ������Gilteritinib�����ҩ���Σ���ר����Ч�ڽ���2030��5�¡�
4.Uliprisnil acetate-NDA������
�ܲ�λ�ڶ����ֵ�Allergan��ҩ��˾�ڵ���ʱ��2017��10��10�շ�������ƣ�FDA�ڽ��������˸ù�˾��ǰ�ύ��Uliprisnil acetate���������ӹ�ƽ��������NDA������ɹ�ͨ����飬Uliprisnil acetate����**���������ӹ�ƽ�������Ŀڷ�ҩ�
Allergan��˾���乫����Ԯ����������������������(AHRQ)�ķ�������ƣ�������Լ��2600��15~50��֮���Ů�Ի����ӹ���������������һ��Ļ����ճ�������ü�����֢״���ܵ�Ӱ�졣�ӹ���������Ҫ֢״���¾���ʧѪ���ࡢ�¾�����**���¾����ڲ�����ƶѪ��������ʹ������ѹ���������֢״�ȡ�
Uliprisnil acetate��һ��ѡ�����м���������ڼ�(SPRM)����ֱ���������ӹ���Ĥ���ӹ�ƽ�����������´����ڵ��м������塣����������������500����������Ů�ԵĢ����ٴ�����(Venus����VENUS��)���Լ�ŷ��������1000�����ӹ�����Ů�Ե�4������Ģ����ٴ������Uliprisnilacetate�İ�ȫ������Ч�Խ�����ϵͳ�����ۡ�
��ǰ��Uliprisnil acetate����ŷ������ô����У���Ʒ���ֱ�ΪEsmya��Fibristal���ֱ���Gedeon Richter��˾��Allergan��˾���ۡ�
Uliprisnil acetate�ṹʽ����ͼ��ʾ��
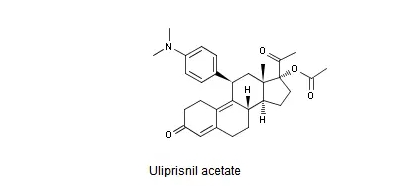
Thomson Reuters Integrity���������ص�Uliprisnil acetate*���ר����US4954490����ͬ��ר����Ȼ����ר��δ�����й���
5.Tebipenem pivoxil-��QIDP��֤
�ܲ�λ���������������ݽ����е�Spero Therapeutics��˾�ڵ���ʱ��2017�귢������ƣ���ǰ�ù�˾���ձ������ƹ���ҩ��ʽ�����SPR994(Tebipenem pivoxil)�Ŀ�������������Եĺϸ�Э�飬Spero Therapeutics��˾ͬʱ������SPR994����FDA�϶�ΪQIDP�������ϸ��Ⱦ������Ʒ��������Ӧ֤�����������������Ⱦ(CUTI)��������(DFI)�����������ϸ���Է���(CABP)��
SPR994�����ձ������ƹ���˾������һ�ֹ��ڷ�̼��ùϩ���ҩ���������ƶ�ҩ��ҩ�Ը�������ϸ�����µĸ�Ⱦ�����а����Է��ŵͪ��ҩ�ĸ�Ⱦ�Լ�������ҩ�����ձ�(2009�꣬��Ʒ��Orapenem)�����������������ۡ�SPR994���ٴ���ҩ���о��У�����Լ1200���������ͯ���߽����˸�ҩ�����ơ�
SPR994�Ļ�ѧ�ṹʽ����ͼ��ʾ��
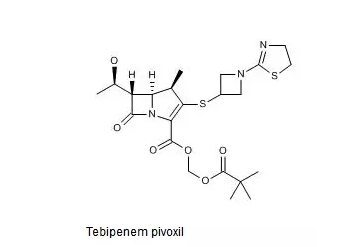
Thomson Reuters Integrity������������SPR994��ص�*���ר����US4644061����ͬ��ר����Ȼ����ר��δ�����й���
6.Pegfilgrastim��������ҩ(Mylan & Biocon)-�յ�FDA��CRL
�ܲ�λ��ӡ���µ����Biocon��ҩ��˾�ڵ���ʱ��2017��10��10�շ�������ƣ�FDA��ǰ�ù�˾��Mylan��ҩ��˾���Ͽ������걨��Pegfilgrastim��������ҩ(MYL-1401H,)�����������ظ���(CRL)����ʱ���������������
Biocon��ҩ��˾��Mylan��ҩ��˾����ȫ��Χ�ھ�6������������Ŀ������к�����MYL-1401H��������֮һ��Biocon��˾�ķ����˱�ʾ���˴�CRL������Ӱ��MYL-1401H�����������мƻ����ù�˾����FDA���й�ͨ���Լ�ʱ���CRL������������⡣
7.Voretigeneneparvovec-��FDA**ίԱ��һ���Ƽ�
�ܲ�λ�������ѳǵ�Spark Therapeutics��˾�ڵ���ʱ��2017��10��12�շ�������ƣ�FDA����֯������Ʒ�����ίԱ������ǰ�����л�������16:0��ͶƱ���ͨ�����飬��ʾ�ù�˾�������걨��������˫��λ����RPE65�鵼���Ŵ�������Ĥ��������ʧ����һ���Ի����Ʒ�LUXTURNA(Voretigeneneparvovec)Ч����ڷ��գ��Ӷ�ʹ��Voretigeneneparvovec������Ϊ��ŵ��CAR-T֮��ĵڶ�����FDA���Ļ����Ʒ���
Luxturna�Ʒ��ɷѳ�Spark Therapeutics��˾���ƣ�ּ������RPE65��������������ļ��������ֱ����Ӱ���۾��Թ�ķ�Ӧ������*�յ�������Ĥ�й�ϵͳʧ�ܡ�
���Ʒ�Ӧ��װ����RPE65��������������IJ�����������ע�䵽�����۾��в�ʹ���л������ṩRPE65������������ס�
��һ���漰31�˵�������������У�Luxturna�ڽ������ƵĻ������϶����ֳ�һ������Ч��SparkTherapeutics����������һ�Ʒ������о���
Thomson Reuters Integrity������ֻ������3����Voretigeneneparvovec�йص�ר����������עΪ����ר���������������ڴ��絽��������US2004022766(δ�����й�)��US2007077228(δ�����й�������һר����Ϊͬ��ר��)��CN201180016616.2(�й�ר����������)��
8.���̫������Ƭ(DSUVIA)-�յ�FDA��CRL
�ܲ�λ�����������������ݵ�AcelRx Pharmaceuticals�ڵ���ʱ��2017��10��12�շ�������ƣ�FDA��ǰ�ù�˾���ύ��DSUVIA(���̫������Ƭ��30mcg)NDA�����������ظ���(CRL)����ʾ��ʱ������NDA��
��Ƭ����ʹҩ���̫��(ҩ������������)�ٴ������ھ�����ӲĤ��������ʹ����Ϊ������ҩ������ó���**�Ӷ����������ٴ�Ӧ�á�AcelRx Pharmaceuticals���������̫��30mcg����Ƭ��ּ�ڿ˷�����ע�������**��ȱ�㣬��ҩ��MAA(����ARX-04)�����ڽ���EMA����顣
AcelRx Pharmaceuticals��ظ����˱�ʾ����ȻFDA��ʱ����DSUVIA�����Ǹ�CRL��������������δ�漰ʵ����ȱ�ݡ�AceRx Pharmaceuticals����FDA�������й�ͨ�����ڼ����ƶ�DSUVIA�����������С�
����Thomson Reuters Integrity���棬���������̫��ע�����1982�꼴�����У�����仯����ר�����ѵ��ڡ�����AcelRx Pharmaceuticals��2012��8��29�շ����Ĺ��棬�ù�˾���DSUVIA������һϵ�е�ר�����ѣ�����*����US8252328��US8252329���漰���̫�ᡣ���⣬espacenet���ݿ�����AcelRx���м����������22��ר���������漰�Ƽ����������IJ����������չ������
9.Apalutamide-�ύNDA
ǿ����ҩ����������ʱ��2017��10��11�շ�������ƣ��ù�˾��ǰ��FDA�ύ����һ���ڷ��ۼ�������(AR)���Ƽ�apalutamide�����������Է�ת����ȥ�Ƶֿ���ǰ���ٰ�(��ת����CRPC)��NDA��ĿǰFDA��δ���κ�ҩ���������Ʒ�ת����CRPC��
ǿ����˾���乫����Ԯ��������֢Э������ݳƣ�2017�������г���16.1�������Ա�ȷ��Ϊǰ���ٰ��������ۼ��ذ�������(ADT)�ķ�ת����ǰ���ٰ�����*�ջ��ADT������ҩ�ԣ��Ӷ���չΪCRPC����������ʾ��10%~20%��ǰ���ٰ�ȷ�ﻼ����5���ڽ�չΪCRPC��
ǿ����˾�Ĺ��滹ָ�����˴��걨�Թؼ��Ԣ����ٴ�����ARN-509-003(SPARTAN)������Ϊ������������������������ADT���ƺ�ǰ�������쿹ԭ(PSA)��ȻѸ�������ķ�ת����CRPC���Ի���Ϊ������ο��Ϊ����������apalutamide����Ч���밲ȫ�ԡ�ǿ����˾��ʾ�������ڽ�����ҽѧ�����Ϲ�����������Ľ����
Apalutamide�Ľṹʽ����ͼ��ʾ��

�й�ר��CN200780019654.7��/��Ȩ˵�����Ȩ��Ҫ��6�Ի�ѧ�ṹʽ����ʽ����������Apalutamide�����ҩ���Σ���ר������2027��3��ʧЧ��
10.Eryaspase-���µݽ�MAA
�ܲ�λ�ڷ����ﰺ��ErytechPharma��˾�ڵ���ʱ��2017��10��10�շ�������ƣ��ù�˾��ǰ��EMA���µݽ�Eryaspase(��Ʒ����Graspa)�������Ʒ����Ի������Լ��Գ��ܰ�ϸ����Ѫ��(ALL)��MAA
Eryaspase����ErytechPharma��������L-���Ŷ�����ø�������˺�ϸ���ڶ��ƵõIJ�Ʒ��2015��9�£��ù�˾��ŷ��EMA�ݽ���Eryaspase��������ALL��MAA����ȴ��ȥ��11�³����˴�MAA������ȡ�㹻��ʱ���ṩCHMP��Ҫ���������Ϣ���˴����µݽ����걨�����а������Զ�ͯ�����ALL����Ϊ����Ģ�/�����ٴ�����GRASPALL2009-06(Clinical Trials.govʶ��ţ�NCT01518517)�Լ�Ϊ�˻ش�CHMP���������Ҫ���������ݡ�GRASPALL2009-06��������ʾ����ԭ̬L-���Ŷ�����ø��ȣ�eryaspase���ϻ��Ƶ����Ʒ����Է����Ի�������ALL������ʾ�����õ���Ч���밲ȫ��������Eryaspase�����黼��L-���Ŷ�����ø���Գ���ʱ�䳤�ȼ�����ԭ̬L-���Ŷ�����ø�����黼�ߵ����������������У�Eryaspase����ʾ�����õİ�ȫ�������������ڽ���eryaspase���ƵĹ����г��ֹ�����Ӧ����ԭ̬L-���Ŷ�����ø�������ڹ�����Ӧ�ķ�����Ϊ48%��eryaspase�������ڻ������յ����ڵ�������ȫ�����ʸ���ԭ̬L-���Ŷ�����ø�����飬��ҩ������Բ�����Ӧ�ķ�����ȴ���ں��ߡ�
��ҩ��δ���κι��һ����С�
Thomson Reuters Integrity����Ԯ��������֢Э������ݳƣ�2017������������6.213����Ѫ��������ÿ�ְ�Ѫ����������������ʾ��
�����ܰ�ϸ����Ѫ��(ALL)��5970��;
�����ܰ�ϸ����Ѫ��(CLL)��2.011����;
��������Ѫ��(AML)��2.318����;
��������Ѫ��(CML)��8950��;
�������͵�Ѫ����5720����
��ҩ��Thomson Reuters Integrity������ֻ������3����֮��ص�ר����
US8617840���й�ͬ��ר��CN200580029814.7��/��Ȩ˵�������Ȩ����ˡ��Ʊ��������Գɷֵĺ�ϸ�����ѽ�/�ٷⷽ��������ר��������Ч���ڣ���Ч�ڽ���2025��8��;
US8974802���й�ͬ��ר��CN200880126046.0����/����˵�������Ȩ��Ҫ���������������ٰ��İ����춬����ø֮��ϸ���Ļ�������������ר���ѱ����أ���ְ�����CN201610012474.7��������;
WO2017114966Ҫ�����õ�����ø���춬����ø���ư�֢�ķ�������δ�����й���
11.Rucaparib-��FDA�ύsNDA
�ܲ�λ���������������ݵ�Clovis Oncology�ڵ���ʱ��2017��10��9�շ�������ƣ��ù�˾��ǰ��FDA�ݽ���Rucaparib������ǰ���ò���ƺ�����ȫ����(CR)�ֻ���(PR)�ķ������ѳ���Ƥ�������ѹܰ���ԭ���Ը�Ĥ������ά�����Ƶ�sNDA����Ϊ�˴�sNDA�걨�����Ģ���˫ä����ο�������ٴ�����(ARIEL3���飬Clinical Trials . govʶ��ţ�NCT01968213)����ļ��564���������ѳ��������ѹܰ���ԭ�����ѳ���Ů�Ի��ߡ������ٴ��������Ҫ��Ч���յ�����ɴӸߵ��͵�3��������Ⱥ���ɣ����ݷ����ڡ���Ҷ�������о����(Coleman RL, et al. ARIEL3 investigators. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017 Sep 12. pii: S0140-6736(17)32440-6.)����ͬ��Ⱥ����λ��չ������(mPFS)������±���ʾ��

��ȫ�Է�����Ⱥ�У�rucaparib���밲ο������3�����������������Բ�����Ӧ�ķ���������Ϊ56%��28%��*�����IJ�����Ӧ��ƶѪ��Ѫ�쵰��Ũ���½�(19% vs 1%)�Լ���������춬����ת��øˮƽ����(10% vs 0)��
Rucaparib��һ�ֶ��ADP-���Ǿۺ�ø(PARP)���Ƽ���ͨ��������PARP-1��PARP-2��PARP-3��DNA�����á�2016��12�£�FDAͨ�����������̶�����Rucaparib 200mg��300mg��250mgƬ(��Ʒ����Rubraca)��Ϊ��һ�Ʒ��������ƽ��ܹ����μ����ϻ��Ƶ�BRCAͻ��������ѳ������ߡ�
Clovis Oncology���乫����Ԯ��������֢Э������ݳƣ�2017������������2.24�����ѳ����������ѳ������䷢�����ڲ��������ɱ��֢״����80~85%���ѳ�����������ȷ��֮ǰ����������ɢ�������������λ�����ɽ������ơ��ѳ������µ������������ڰ�֢���µ������������������壬�Ҹ�������������ֳϵͳ��֢��
Rucaparibҩ�����������Ի�����(camsylate)���ṹ����ͼ��ʾ��

�й�ר��CN00804589.5��/��Ȩ˵�����Ȩ��Ҫ��3�Ի�ѧ�ṹʽ����ʽ�����˰���Rucaparib���ڵĶ��������PARP���Ƽ������ҩ���Σ���ר����Ч�ڽ���2030��1�¡�
�й�ר��CN201180009237.0��/��Ȩ˵�����Ȩ��Ҫ��1�Ի�ѧ������ʽ������Rucaparib���Ի����Σ�Ȩ��Ҫ��3��4�ֱ���������Ի����εĹ���������������ר������Ч�ڽ���2031��2�¡�
�� �ٴ�����
1.PC-mAb������a���ٴ�����
�ܲ�λ����˹�¸��Ħ��Athera Biotechnologies AB��˾�ڵ���ʱ��2017��10��11�շ�������ƣ��ù�˾������һ������PC-mAb�Զ���Ѫ����֢���������õ�ǰհ�ԡ�˫ä�����������ο�����յĶ������ٴ����顣�����������������Ⱥ��֬����a(Lp(a))ˮƽ���ߵĻ��߹��ɣ�������֬����a����֬������(PC)�н�ǿ��Я����������PC����PC-mAb����֢��Ӧ�����ưе㡣
2.Tinostamustine������/�����ٴ�����
�ܲ�λ����ʿ��MundipharmaEDO��˾�ڵ���ʱ��2017��10��10�շ�������ƣ��ù�˾����������������tinostamustine(EDO-S101)��������ʵ�������ߵĢ�/�����ٴ����顣����������ڲ��ֵ���ҪĿ��������tinostamustine������ҩʱ�İ�ȫ�ԡ������ԡ�*�����ܼ�����Ϊ�����ٴ�����ɸѡ����������������������ҩ������ѧ������Ϊ���ҪĿ�ģ��������黹��̽���Կ�����п��������Ե����������л������ı仯�̶ȣ�������ڲ��ֵ�����������Сϸ���ΰ�������֯��������KITθ�������������������ٰ������Ⱦ���ʵ�����Ļ��ߡ�
Tinostamustine��һ��������ø���Ƽ����黯�����仯ѧ�ṹʽ����ͼ��ʾ��
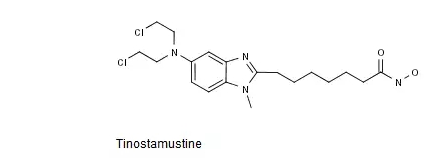
�й�ר��CN201080002890.X����Ȩ˵�����Ȩ��Ҫ��3�Ի�ѧ�ṹʽ����ʽ����������Tinostamustine�����ҩ���Σ���ר����Ч�ڽ���2030��1�¡�
�� ��ҵ��Ϊ
1.KVD-001-����Ȩת��
�ܲ������������������ݽ����е�KalVista��˾�ڵ���ʱ��2017��10��10�շ������棬Ĭ��Merck & Co(NYSE:MRK)�Ѿ�����910����Ԫ(8.50��Ԫ/��)ȡ�øù�˾9.9%������Ȩ��
���⣬Ĭ�˻���֧��KalVista 3700����ԪԤ�������չ��������Իư�ˮ��ѡҩ����Ȩ�����ҹ�˾������������ҩ����⣬Ĭ�˻����δ�������ڷ������Իư�ˮ�������������Ȩ��KalVista���п��ܻ��*��7.15����Ԫ�Ķ���֧����Լ����ھ����۶�ķֲ�ר��Ȩ���ɷѡ�
KalVista�ƻ���չ�����Իư�ˮ������ҩ��KVD001�Ķ������顣�����Իư�ˮ����һ��������֢�����ܵ������������ʧ����KalVista�ɼ���һ���̱�7.35��Ԫ����ȥ����������0.3%��ͬ�ڱ���500ָ��(SPX)����4.8%��Merck�ɼ���ǰ����0.5%��
KVD-001��һ��Ѫ�������ͷ�ø���Ƽ�����ʱδ�鵽����ṹʽ�Լ�ר���йص���Ϣ��
2.INB03-����Ȩת��
�ܲ�λ�����������������ݵ� INmune Bio ��˾�ڵ���ʱ��2017��10��10�շ�������ƣ��ù�˾��ǰ�� Xencor Inc. ��˾�����**���������Ƽ�INB03�Ŀ���Ȩ������ָ�����ְ�֢����**����ϵͳ����Դ����ϸ��(MDSC)ˮƽ������������ѪҺ�����������ڵ�MDSCˮƽ������Ԥ�⼲�������س̶ȣ���֢�����������Լ��������м��վ���Ƽ����ڵ����������Ʒ�����ʧ�ܵĿ����ԡ�MDSC�����ڵ�����������ϸ��������ʹ����������������ϵͳ�Ĺ�����������MDSC�����Ч���ƶ���֢���Ƶķ�չ.
���α༭���ɺ� WWW.1168.TV 2017-10-19 11:40:36
������Դ��
1.������ע������Դ��1168ҽҩ����������������Ʒ����Ϊ���ݽ��ڻ������Ƽ�����˾-1168ҽҩ�������Ϸ�ӵ�а�Ȩ����Ȩʹ�õ���Ʒ��δ��������Ȩ����ת�ء�ժ�������������ʽʹ��������Ʒ���Ѿ�������Ȩʹ����Ʒ�ģ�Ӧ����Ȩ��Χ��ʹ�ã���ע������Դ��1168ҽҩ������http://www.1168.tv����Υ�����������ߣ�������������ط������Ρ�
2.����ת�ز�ע����������Դ����1168ҽҩ������������Ʒ��Ŀ�����ڴ��ݸ�����Ϣ����������������ͬ��۵��Ͷ�����ʵ�Ը��𣬲��е�������Ʒ��Ȩ��Ϊ��ֱ�����μ��������Ρ�
3.����ý�塢��վ����˴ӱ���ת��ʱ�����뱣������ע������Ʒ��һ��Դ�����Ը���Ȩ�ȷ������Ρ�
4.���漰��Ʒ���ݡ���Ȩ�����⣬������Ʒ����֮����һ�����뱾����ϵ��������Ϊ�������Ȩ������ϵ���䣺1753418380@qq.com��

�����÷�Χ�����ڻ��⾱���硢�����ȼ��պ�������֯��ʹ�����͵Ȳ���֢״��Ⱥ���������ʹ�÷��������á�����Ʒ����ֱ��ͿĨ�ڲ��ʲ�λ�����ᰴĦ2-3���ӣ�ÿ��2-3�Ρ�

�����÷�Χ�����ڻ��⾱���硢�����ȼ��պ�������֯��ʹ�����͵Ȳ���֢״��Ⱥ���������ʹ�÷��������á�����Ʒ����ֱ��ͿĨ�ڲ��ʲ�λ�����ᰴĦ2-3���ӣ�ÿ��2-3�Ρ�








 ���������� 44011102000390��
���������� 44011102000390��






